
Destylacja pod ciśnieniem atmosferycznym
Typowy zestaw do oczyszczania związków ciekłych za pomocą zwykłej destylacji pod ciśnieniem atmosferycznym przedstawiono na rysunku 1.Kolba destylacyjna może mieć każda, odpowiednią do ilości cieczy pojemność, chociaż trzeba dodać, że do małych ilości (od 3 do 25 ml) lepiej użyć kolby gruszkowej. Wymiary kolby powinny być tak dobrane, aby substancja przeznaczona do destylacji zajmowała od połowy do dwóch trzecich objętości kolby. Nagwintowany łącznik z nakrętką umieszczony w nasadce destylacyjnej umożliwia umiejscowienie kulki termometru nieco poniżej poziomu bocznej rurki nasadki. Gdy temperatura wrzenia cieczy przekracza 150°C, chłodnicę wodną zastępuje się długą rurką (bez płaszcza) ze szlifami przy obydwóch końcach, działającą jako chłodnica powietrzna. W przypadku, gdy destylat trzeba zabezpieczyć przed wilgocią powietrza, do bocznej rurki przedłużacza przyłącza się rurkę osuszającą napełnioną bezw. chlorkiem wapnia, utrzymywanym w rurce za pomocą zatyczek z waty. Do kolby napełnionej cieczą dodaje się kilka kawałków porowatej nie polewanej porcelany, tzw. porcelanki, zapewniających równomierne wrzenie cieczy podczas ogrzewania. W żadnym przypadku nie wolno wrzucać porcelanki do gorącej cieczy gdyż może to spowodować wyrzucenie zawartości kolby na zewnątrz. Kolbę ogrzewa się za pomocą czaszy lub lepiej w łaźni odpowiedniej do ogrzewania do temperatury wrzenia cieczy poddawanej destylacji. Kolby gruszkowe z małymi ilościami cieczy można ogrzewać na mniejszej czaszy grzejnej. Dopóki nie rozpocznie się wrzenie cieczy, ogrzewa się kolbę raczej intensywnie, następnie zmniejsza się intensywność ogrzewania i reguluje dopływ ciepła tak, aby destylat spływał z chłodnicy z szybkością l-2 krople na sekundę. Należy dodać, że na początku destylacji ogrzanie górnej części kolby i termometru przez pary związku wymaga pewnego czasu. Destylacja nie powinna być prowadzona zbyt powoli, ponieważ termometr oziębia się w przypadku niedostatecznego dopływu świeżych par do zbiornika z rtęcią i odczyty temperatury są nieprawidłowe
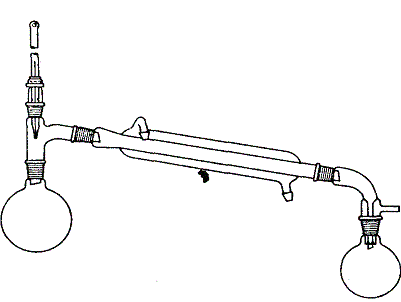
Rys.1 Zestaw do destylacji pod ciśnieniem atmosferycznym
Po dojściu cieczy do stanu wrzenia, obserwuje się na termometrze najpierw szybki wzrost temperatury, następnie, w pobliżu temperatury wrzenia, wzrost ten staje się powolny i wreszcie temperatura praktycznie ustala się. W tym momencie do aparatury przyłącza się czysty zważony odbieralnik i zbiera destylat tak długo, aż w kolbie destylacyjnej pozostanie tylko mała objętość cieczy. W regularnych odstępach czasu należy notować wskazania termometru. Jeżeli destylowana ciecz nie jest zbyt zanieczyszczona, to większość jej powinna przed destylować w wąskich granicach temperatury w przedziale 2-3°C, gdy temperatura nie ustala się, lecz stale wzrasta, oznacza to, że związku nie można oczyścić za pomocą destylacji zwykłej i że trzeba zastosować destylację frakcyjną. Do destylacji cieczy w ilości od 0,5 do 3,0 ml dostępne są kolby gruszkowe o pój. 2 ml i 5 ml (wymiar szlifu 7/11) z przytwierdzoną do bocznego ramienia chłodniczką (rys.2).

Rys. 2. Kolba z przytwierdzona chłodnicą do destylacji małychilości cieczy
Zaleta tego rodzaju zestawu polega na zmniejszeniu strat wynikających z zatrzymania cienkiej warstewki destylatu na powierzchni szklanej.
Boczne ramię ma u wylotu specjalną oszlifowaną końcówkę ułatwiającą ściekanie i zbieranie destylatu. Kolbę ogrzewa się starannie regulując
dopływ ciepła.
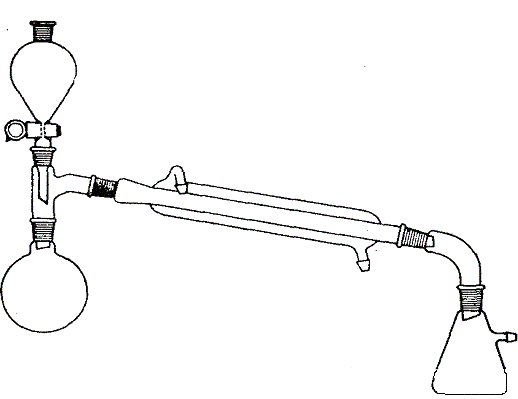
Rys. 3. Zestaw do oddestylowania
Zestaw przedstawiony na rys. 3 stosowany jest do oddestylowania rozpuszczalnika z roztworu otrzymanego np. w wyniku ekstrakcji. Kolbę napełnia się do potowy roztworem; pozostały roztwór wlewa się do wkraplacza i wkrapla go do kolby z taką samą w przybliżeniu szybkością, z jaką destyluje i spływa do odbieralnika rozpuszczalnik. Zbędne jest, więc użycie dużej kolby mieszczącej cały rozpuszczalnik oczyszczanie wielu wysoko wrzących substancji przez destylację w stosunkowo niskiej temperaturze jest to szczególnie cenne, gdy związek organiczny rozkłada się w czasie destylacji pod ciśnieniem atmosferycznym. Destylacja z parą wodną ma też duże znaczenie jako sposób oddzielania pożądanego związku organicznego w następujących przypadkach:
Destylacja frakcyjna pod ciśnieniem atmosferycznym
Jeżeli różnica w temperaturach wrzenia składników mieszaniny, nie jest odpowiednio duża, to przy ich rozdzielaniu za pomocą destylacji stosuje się na ogół kolumnę destylacyjną i frakcyjną. Aparatura do precyzyjnego frakcjonowania, za pomocą, której można z powodzeniem rozdzielić mieszaninę składników o temperaturach wrzenia różniących się zaledwie o kilka stopni, znajduje się w sprzedaży, lecz dla uzyskania dobrych wyników niezbędne jest również staranne wykonanie destylacji frakcyjnej i umiejętność doceniania czynników wywierających wpływ na sprawność kolumny destylacyjnej. Kolumna destylacyjna jest to długa pionowa rura, poprzez którą pary podążają ku górze, ulegając w tym czasie częściowemu skropleniu; skropliny spływają w dół kolumny i są zawracane do kolby. Wewnątrz kolumny następuje bezpośrednie zetknięcie się zawracanej cieczy z dążącymi ku górze parami. W wyniku wymiany cieplnej pomiędzy dwiema fazami dążącymi do stanu równowagi, pary zostają wzbogacone w bardziej lotny składnik kosztem fazy ciekłej. Dla dobrego rozdzielenia destylowanej mieszaniny niezbędne są następujące warunki:
1) stosunkowo duża ilość cieczy zawracanej w kolumnie,
2) dokładne wymieszanie się fazy ciekłej i gazowej,
3) duża aktywna powierzchnia zetknięcia się obydwu faz.
Należy unikać nadmiernego ochłodzenia kolumny; dotyczy to zwłaszcza destylacji cieczy o wysokich temperaturach wrzenia. Przeciwdziała temu skutecznie izolowanie zewnętrznej powierzchni kolumny i zastosowanie płaszcza próżniowego lub ogrzewania elektrycznego. Na rysunku 1 przedstawiono zestaw do zwykłej destylacji frakcyjnej z zastosowaniem kolumny Vigreux. Kolumna ta, o umiarkowanej sprawności, jest prawdopodobnie jedną z najczęściej stosowanych kolumn. Kolumnę Vigreux stanowi rura szklana z wgięciami wykonanymi w ten sposób, że co drugie wgięcia na tym samym poziomie są skierowane w dół pod kątem 45°. Ma to na celu zbieranie cieczy ze ścian i przeniesienie jej do środka kolumny. Mieszaninę, którą poddaje się destylacji, umieszcza się w kolbie o takiej pojemności, aby ciecz wypełniła trzecią część lub połowę kolby, i wrzuca się kilka kawałków porowatej porcelany. Boczną rurkę kolumny łączy się z chłodnicą wodną. Destylat zbiera się w małych kolbkach stożkowych lub w probówkach

Rys. 1 Zestaw do destylacji frakcyjnej z kolumną Vigreux
Zbiornik termometru powinien znajdować się nieco poniżej poziomu bocznej rurki kolumny. Celowe jest owinięcie kolumny tkaniną lub folia aluminiowa owiniętą materiałem; ogranicza to ochładzanie kolumny prądem powietrza. Izolowanie kolumny ma zasadnicze znaczenie wtedy, gdy którykolwiek ze składników mieszaniny wrze w temperaturze przekraczającej 100°C. Kolbę ogrzewa się na łaźni powietrznej lub olejowej umożliwiającej równomierne ogrzewanie. Początkowo nie można ogrzewać zbyt szybko, gdyż następuje wtedy intensywne skraplanie się cieczy w nieogrzanej kolumnie i tym samym zalewanie kolumny cieczą. Gdy destylacja już rozpocznie się, dopływ ciepła reguluje się w ten sposób, żeby w ciągu 2-3 s przechodziła jedna kropla destylatu. W tych warunkach powinno się uzyskać dość wydajne rozdestylowanie mieszaniny. Jeśli kolumna destylacyjna zdolna jest do wyraźnego rozdestylowanie mieszaniny na poszczególne frakcje, to po przedestylowaniu frakcji nisko wrzącej destylacja ustaje. Wtedy zwiększa się powoli ogrzewanie, aż zacznie destylować druga frakcja, czemu towarzyszy znaczny wzrost temperatury odczytanej na termometrze. W przypadku mało sprawnej kolumny otrzymuje się stosunkowo znaczne ilości frakcji pośrednich. Należy podkreślić, że destylację frakcyjną trzeba prowadzić powoli; destylując szybko nie zaoszczędza się czasu, gdyż zazwyczaj konieczna jest destylacja powtórna. Kolumna napełniona jest odpowiednim wypełnieniem sięgającym o 5 cm poniżej szczytu kolumny. Wypełnienie opiera się na małej szklanej spirali o odpowiednich wymiarach. W sprzedaży znajduje się kilka rodzajów doskonałych wypełnień kolumny. Najprostszym, najtańszym i jednocześnie bardzo skutecznym wypełnieniem są szklane pierścienie o wysokości 6-9 mm i średnicy 6-9 mm, zwane pierścieniami Raschiga (rys. I,91a); podobne pierścienie porcelanowe są prawie tak samo skuteczne. Porcelanowe pierścienie Lessinga (rys. I,91b) są to puste wewnątrz cylindry o jednakowej wysokości i średnicy; w środku cylindra znajduje się przegródka. W metalowym pierścieniu Lessinga (rys. I,91c, dobre wyniki uzyskuje się z pierścieniami aluminiowymi, miedzianymi i niklowymi, jeżeli nie zachodzi oddziaływanie chemiczne pomiędzy metalem i parami cieczy) boczna powierzchnia
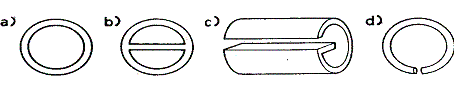
Rys. 1,91. Wypełnienie kolumn destylacyjnych: a) pierścienie Raschiga, b) porcelanowe pierścienie Lessinga, c) metalowe pierścienie Lessinga, d) pierścienie Fenskego
cylindra nie jest pełna, a przegródka skierowana wzdłuż średnicy jedną stroną łączy się z cylindrem, a drugą nie dotyka ścianki cylindra. Zastosowanie omawianej przegródki w cylindrach zwiększa sprawność wypełnienia, gdyż powstaje w ten sposób dodatkowa powierzchnia zetknięcia par z cieczą. Obydwa wymienione rodzaje wypełnienia są stosunkowo tanie. O ich sprawności świadczy destylacja frakcyjna mieszaniny 50 ml benzenu (tw. 80°C) i 50 ml toluenu (tw. 110°C) z użyciem kolumny Hempla wypełnionej porcelanowymi pierścieniami Raschiga lub aluminiowymi pierścieniami Lessinga (przy wypełnieniu kolumny o dług. 35 cm i średnicy 18 mm). W wyniku destylacji otrzymuje się 43-47 ml czystego benzenu i 44-46 ml czystego toluenu. Jednozwojowe szklane heliski, zwane pierścieniami Fenskego (rys. I,91d) stanowią również, aczkolwiek nieco kosztowniejsze, wypełnienie kolumny destylacyjnej Hempla. Dogodne są heliski o zewn. średnicy poszczególnego zwoju 4,0 mm i o grubości pałeczki 0,5 mm; l kg helisków zajmuje w przybliżeniu objętość 2 l. Heliski te tworzą ściśle ułożone wypełnienie zapewniające maksymalną powierzchnię zetknięcia pomiędzy Stale spływającą cieczą i strumieniem par i jednocześnie umożliwiają dobry przepływ cieczy i par. Szczegółowe omówienie teorii destylacji frakcyjnej przekracza zakres podręcznika, zostaną tylko podane użytkowane pojęcia oraz zasadnicze wymagania dotyczące sprawności kolumny destylacyjnej. Pojemność kolumny odpowiada ilości par i cieczy, które płyną w kolumnie przeciwprądowo, nie powodując zalewania kolumny cieczą. Sprawność kolumny określa się zdolnością frakcjonowania odcinka kolumny o określonej długości; oznacza się ją, porównując zdolność frakcjonowania kolumny z obliczoną zdolnością frakcjonowania teoretycznie doskonałej kolumny o jednej półce w takich samych warunkach. Pólka teoretyczna określa sekcję kolumny destylacyjnej o takiej długości, na jakiej pary uchodzące z półki mają ten sam skład, co pary, które w tej samej temperaturze pozostają w równowadze z cieczą według wykresu para faza ciekła. Ponieważ sprawność kolumny zależy od ustalania się warunków równowagi pomiędzy dążącymi ku górze parami i spływającą w dół cieczą na skutek dokładnego ich wymieszania, jest oczywiste, że bardziej lotny składnik powinien być odbierany ze szczytu kolumny możliwie jak najwolniej. Liczby półek teoretycznych nie można określić na podstawie wymiarów kolumny; oblicza się ją na podstawie wyników rozdestylowania mieszanin wzorcowych, dla których skład par i cieczy jest dokładnie znany (np. benzen i toluen, benzen i czterochlorek węgla, benzen i dichloroetan, heptan i metylocykloheksan). Zwykła rura o średnicy l cm i dług. l m odpowiada tylko jednej półce teoretycznej, podczas gdy ta sama rura z odpowiednim wypełnieniem może osiągnąć sprawność 20 lub więcej półek teoretycznych. Kolumna o 12 półkach teoretycznych praktycznie wystarcza do rozdzielenia benzenu i toluenu (A tw. 30°C), lecz gdy składniki mieszaniny mają temp. wrzenia różniące się zaledwie o 3°, to do ich rozdestylowania wymagana jest kolumna o ok. 100 półkach teoretycznych. Działanie kolumny zależy zarówno od wysokości i wypełnienia kolumny, jak i od jej konstrukcji i dlatego sprawność kolumny wyraża się często wysokością równoważna półce teoretycznej. Oblicza się ją dzieląc wysokość kolumny przez liczbę półek teoretycznych i określa się ją zazwyczaj w centymetrach. Porównywanie względnych sprawności kolumn destylacyjnych należy przeprowadzić w warunkach standardowych. Idealna destylacja frakcyjna daje wyraźne frakcje wrzące w określonym i wąskim zakresie temperatury. Po przedestylowaniu każdej frakcji temperatura wzrasta gwałtownie, przy czym nie destyluje frakcja pośrednia. Jeśli sporządzi się wykres zależności temperatury wrzenia od objętości destylatu, to w przypadku idealnej destylacji frakcyjnej otrzyma się linie proste, kolejno poziome i pionowe, przypominające schodki. Mniej lub bardziej pochyłe załamanie linii schodkowej wskazuje na obecność frakcji pośredniej. Ilość tej frakcji pośredniej stanowi jakościowe kryterium działania różnych kolumn. Zasadniczym celem przy projektowaniu różnych kolumn destylacyjnych jest zmniejszenie ilości frakcji pośrednich do minimum. Do najważniejszych czynników wpływających na skuteczność rozdzielania mieszanin na frakcje wrzące w wąskim zakresie temperatury należą następujące:
1. Czas destylacji. Każdej kolumnie odpowiada zawsze pewien optymalny czas destylacji. Skrócenie tego czasu powoduje zmniejszenie się dokładności rozdestylowania mieszaniny, natomiast nadmierne przedłużenie czasu destylacji nie jest racjonalne, chociaż zapewnia nieco lepsze rozdestylowanie. Destylacja frakcyjna z zastosowaniem kolumn laboratoryjnych trwa od l h do 8-10 h.
2. Orosienie kolumny. Ilość cieczy określana nazwą orosienia kolumny powinna być zmniejszona do minimum zapewniającego dostateczną skuteczność przemywania oparów; powinna też być dostosowana do pojemności kolumny. Stosunek ilości cieczy wprowadzonej do aparatu destylacyjnego do ilości cieczy zraszającej kolumnę powinien być możliwie największy; na ogół ilość cieczy przeznaczonej do destylacji powinna być, co najmniej dwudziestokrotnie większa od ilości zraszającej kolumnę.
3. Izolacja cieplna. Nawet niewielkie straty cieplne powodują znaczne przesunięcie stanu równowagi kolumny. W przypadku, zatem mieszaniny, której składniki różnią się temperaturami wrzenia zaledwie o kilka stopni, rozdzielenie ich wymaga zastosowania kolumny o prawie doskonałej izolacji cieplnej. Teoretycznie największą sprawność kolumny osiąga się w warunkach adiabatycznych. Jeżeli składniki mieszaniny wrą poniżej 100°C, wystarcza zastosowanie próżniowego posrebrzanego płaszcza, przy czym sprawność płaszcza zależy od staranności jego wykonania: oczyszczenia, posrebrzenia i usunięcia z niego powietrza. Na ogół najbardziej celowym sposobem izolowania kolumny jest dostarczanie ciepła równoważącego straty cieplne. W tym celu kolumnę umieszcza się w płaszczu ogrzewanym elektrycznie, przy czym temperaturę płaszcza reguluje się za pomocą opornicy albo autotransformatora z ciągłą regulacją (typu Variac). Temperatura płaszcza powinna być o 5°C niższa od temperatury par skraplających się w górnej części kolumny. Płaszcz ogrzewany elektrycznie przygotowuje się w łatwy sposób z dwóch szklanych rur pyreksowych. Długości tych rur dobiera się w taki sposób, żeby płaszcz umieszczony tuż powyżej dolnego końca kolumny, zakończonego szlifem, sięgał aż do dolnej części głowicy. Wewnętrzna rura ma średnicę 35 mm, a zewnętrzna 55 mm. Kolumnę z przymocowanym termometrem umieszcza się w rurze wewnętrznej (rys. 1,93). Na węższą rurę nawija się elektryczną spiralę grzejną o średnicy zwojów 20 mm, regulując ogrzewanie opornicą, regulatorem mocy lub autotransformatorem. Końce płaszcza zamyka się zatyczką azbestową lub inną zatyczką izolującą o odpowiedniej wielkości i odpowiednim kształcie.
4. Stopień deflegmacji. Stopniem deflegmacji nazywa się stosunek ilości cieczy zawracanej do kolumny (w molach) do ilości destylatu (w molach) odbieranej w jednostce czasu. W zależności od trudności rozdestylowania mieszaniny należy odpowiednio zmieniać stopień deflegmacji. Chcąc osiągnąć dużą sprawność kolumny, trzeba dobrać duży stopień deflegmacji. Wyrażając to inaczej, można przyjąć, że jeśli zmniejsza się stopień deflegmacji, (czyli zmniejsza się ilość cieczy zawracanej do kolumny, a wzrasta ilość destylatu), to liczba półek teoretycznych wymagana do rozdestylowania mieszaniny zwiększa się. Zmianę stopnia deflegmacji osiąga się za pomocą odpowiedniej głowicy, zwykle głowicy do całkowitej kondensacji, z regulowanym odpływem destylatu. W głowicy tej cała para ulega skropleniu, część kondensatu zostaje zawrócona do kolumny, a pozostała część jest zbierana w odbieralniku. Jedną z dostępnych w handlu głowic przedstawiono na rys. 1,92, inne zilustrowano w zestawach do destylacji opisanych poniżej. Rysunek 1,93 przedstawia zdjęcie ogólnie przydatnego zestawu do destylacji frakcyjnej z zastosowaniem szklanych helisków jako wypełnienia. Kolumna zaopatrzona jest w płaszcz ogrzewany elektrycznie, którego temperaturę nastawia się za pomocą autotransformatora z regulacją ciągłą. Zestaw zaopatrzony jest w głowicę do całkowitej kondensacji z regulowanym odpływem destylatu. Na szczycie kolumny całość pary ulega skropleniu. Przez odpowiednie nastawienie specjalnego kranu (umożliwiającego dokładne dobranie stopnia deflegmacji) część skroplonej cieczy zawraca się do kolumny, resztę zaś odprowadza do odbieralnika Korzyści związane ze stosowaniem głowicy polegają na tym, że można stworzyć warunki pełnej równowagi przed odbiorem destylatu, co jest szczególnie ważne wtedy, kiedy należy odpowiednio nastawić temperaturę płaszcza grzejnego. Poza tym, przejście od frakcji nisko wrzącej do frakcji wrzącej wyżej jest
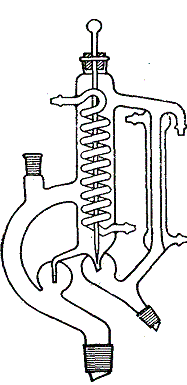

Rys. 1,92. Głowica do całkowitej kondensacji z regulowanym odpływem kondensatu Rys. 1,93. Zestaw do destylacji frakcyjnej z wypełnieniem ze szklanych helisków
stosunkowo łatwe. Ciecz skrapla się przy zamkniętym kranie tak długo, aż termometr wskaże temperaturę najniżej wrzącej frakcji. W tym momencie kolumna osiąga największy stopień rozdestylowania mieszaniny i ustala się w niej równowaga. Wtedy otwiera się częściowo kran i zbiera destylat. Gdy temperatura zacznie wzrastać, zamyka się kran i znów doprowadza do stanu równowagi w kolumnie, po czym zbiera się następną frakcję. Postępując w ten sposób osiąga się dokładniejsze rozdzielenie mieszaniny na ostro wrzące frakcje. Dalsze ulepszenie polega na odprowadzaniu destylatu do odbieralnika za pomocą rurki kapilarnej. Stopień deflegmacji określa się w przybliżeniu, porównując liczbę kropli zawracanej do kolumny cieczy z liczbą kropli cieczy spływającej do odbieralnika (krople spływające ze skośnie ściętych końców rurek łatwo jest zaobserwować. W aparacie można przeprowadzić destylację frakcyjną pod zmniejszonym ciśnieniem wykorzystując nasadkę do destylacji „próżniowej", również pokazaną na rys. 1,93. Destylację frakcyjną przeprowadza się w sposób następujący:
1. Kolbę napełnioną cieczą łączy się z kolumną. Głowicę destylacyjną nastawia się na całkowite zawracanie kondensatu i ogrzewa kolbę tak, aby w kolumnie zaczęły skraplać się pary. Następnie ogrzewa się kolumnę bardzo wolno, aż skraplające się pary dojdą do szczytu kolumny i termometr wskaże temperaturę wrzenia. Wtedy temperaturę płaszcza (lub górnej jego części), rejestrowaną na termometrze przylegającym do kolumny, nastawia się w taki sposób, żeby była o 5°C niższa od odczytanej temperatury wrzenia. Jeśli w kolumnie jest wypełnienie ułożone bezładnie, jak np. szklane heliski lub pierścienie z siatki, kolumna powinna najpierw zostać zalana, tak żeby całe wypełnienie zostało całkowicie zwilżone cieczą. Stan całkowitego zawracania kondensatu utrzymuje się, zatem do chwili ustalenia się równowagi (ok. l h przy 10 półkach teoretycznych).
2. Gdy w kolumnie ustali się równowaga, głowicę nastawia się na pożądany stopień deflegmacji, następnie zmienia odbieralnik i zbiera najniżej wrzącą frakcję w zakresie l-2°C. Podczas destylacji szybkość zawracania kondensatu do kolumny powinna, być możliwie największa, jednak dobrana w taki sposób, żeby kolumna nie była zalewana cieczą. Zachowując takie warunki, dobiera się stopień deflegmacji przez odpowiednią regulację szybkości zbierania destylatu. W miarę usuwania najniżej wrzącego składnika mieszaniny ilość jego w kolbie destylacyjnej zmniejsza się i do szczytu kolumny może dojść mieszanina dwóch składników, przy czym temperatura wrzenia nieco wzrasta. Wtedy należy stopniowo zwiększać stopień deflegmacji, tzn. zmniejszać szybkość zbierania się destylatu; w ten sposób umożliwia się zebranie najniżej wrzącego składnika w wąskim zakresie temperatur. Jednak po upływie pewnego czasu nawet przy dość dużym stopniu deflegmacji temperatura wrzenia wzrasta. Wówczas zmienia się odbieralnik i zbiera frakcję pośrednią.
3. Podczas destylacji frakcji pośredniej destylat należy zbierać bardzo powoli. Temperatura wrzenia wzrasta, a potem ustala się, albo też rośnie bardzo powoli. Wtedy zmienia się odbieralnik, regulując znów temperaturę płaszcza grzejnego i zbiera w wąskim zakresie temperatur drugą frakcję destylatu początkowo szybko, dopóki utrzymuje się stała temperatura wrzenia, a następnie wolniej. Wreszcie, gdy zacznie destylować druga frakcja pośrednia, zmienia się odbieralnik i znów zbiera się frakcję pośrednią bardzo wolno. Następnie postępuje się tak, jak poprzednio, zbierając trzeci składnik mieszaniny i dalsze. Konieczne są następujące wyjaśnienia dotyczące szczegółów postępowania podczas destylacji frakcyjnej:
a) Im lepsze jest rozdestylowanie mieszaniny na ostro wrzące frakcje, tym mniejsze są ilości frakcji pośrednich. Jeśli temperatury wrzenia składników mieszaniny różnią się znacznie, to rozdestylowanie mieszaniny jest tak łatwe, że praktycznie cała ilość niżej wrzącego składnika przechodzi w stałej temperaturze wrzenia. Po pewnym czasie pary przestają dochodzić do górnej części kolumny, szybkość destylacji zmniejsza się i wreszcie destylacja ustaje, a u dołu kolumny zachodzi silne skraplanie. Temperatura par zaczyna obniżać się, aż wreszcie spada poniżej temperatury górnej części płaszcza ogrzewającego. Zwykłe podwyższenie temperatury łaźni ogrzewającej kolbę wywołuje w takim przypadku zalewanie kolumny cieczą; należy, więc stopniowo zwiększać ogrzewanie płaszcza, tak żeby skraplające się pary osiągnęły znowu szczyt kolumny, wtedy temperatura wrzenia zacznie wzrastać, a po pewnym czasie ustali się; temperaturę płaszcza utrzymuje się nieco poniżej temperatury wrzenia.
b) Gdy pod koniec destylacji pewnej frakcji zmniejsza się szybkość zbierania destylatu, należy również zmniejszyć nieco temperaturę łaźni ogrzewającej, a to w celu uniknięcia zalewania kolumny cieczą. Kiedy podczas destylacji frakcji pośredniej temperatura wrzenia wzrasta, należy zwiększyć ogrzewanie płaszcza, tak żeby temperatura jego była tylko nieco niższa od temperatury wrzenia.
c) Jeśli u dołu kolumny pary skraplają się tylko w nieznacznym stopniu, a u szczytu kolumny następuje zalewanie cieczą, wskazuje to, że temperatura płaszcza ogrzewającego kolumnę jest za wysoka. Gdy natomiast u dołu kolumny występuje normalne silne skraplanie, a szczyt kolumny ulega zalaniu cieczą, to prawdopodobnie temperatura łaźni ogrzewającej jest za wysoka. Jeśli dół kolumny jest zalewany cieczą, a u szczytu kolumny pary skraplają się tylko w nieznacznym stopniu, wskazuje to, że temperatura płaszcza jest za niska
d) W przypadku gdy pożądane jest przedestylowanie cieczy pozostającej w kolumnie po zakończeniu destylacji (stanowiącej orosienie kolumny), przed rozpoczęciem destylacji do kolby należy dodać cieczy wyżej wrzącej w ilości nieco większej od ilości stanowiącej orosienie kolumny. Temperatura wrzenia dodanej cieczy powinna być, co najmniej o 20°C wyższa od końcowej temperatury wrzenia destylowanej mieszaniny. Oddestylowując końcową frakcję mieszaniny, reguluje się temperaturę łaźni ogrzewającej kolbę w taki sposób, żeby najwyżej wrzący składnik mieszaniny destylował, a płaszcz ogrzewa się powoli i ostrożnie do temperatury nieco wyższej od temperatury wrzenia tego składnika. Dodana do mieszaniny destylowanej ciecz powinna być chemicznie bierna, tania i nie powinna dawać mieszanin azeotropowych ze składnikami mieszaniny; stosuje się do tego celu następujące ciecze: toluen, tw. 110°C; p-cymen, tw. 175°C; tetrahydronaftalen, tw. 207°C i eter difenylowy, tw. 259°C
Kolumna z wirującą wstęgą
Mieszaninę składników o temperaturach wrzenia różniących się zaledwie o kilka stopni można z powodzeniem rozdestylować za pomocą kolumny z wirującą wstęgą.
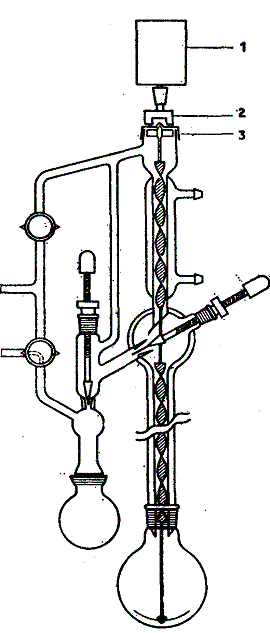
Kolumnę destylacyjną stanowi pionowa rura szklana, w której umieszczona jest wzdłuż całej długości rury dokładnie dopasowana spirala z teflonu lub siatki metalowej (z platyny, stali nierdzewnej lub stopu Monela), przymocowana do teflonowego lub metalowego pręta, średnica spirali jest bardzo niewiele mniejsza od wewnętrznej średnicy rury. Spirala przechodzi również przez chłodnicę spełniającą rolę głowicy do całkowitej kondensacji z regulowanym odbiorem destylatu. Spiralę wprowadza się w ruch obrotowy za pomocą albo bezpośredniego, albo magnetycznego połączenia z silnikiem elektrycznym (rysunek powyżej Centralny pręt wirującej wstęgi sięga do kolby destylacyjnej, gdzie przechodzi w mieszadło teflonowe, zapewniające równomierne wrzenie. Kolumnę umocowuje się w płaszczu z chromoniklowym elementem grzejnym, albo umieszcza się ją wraz z głowicą w posrebrzanym płaszczu próżniowym. Wirująca wstęga, (która obraca się z wybraną prędkością pomiędzy 600 i 3000 obr/min) rzuca pary destylowanej mieszaniny na ściany kolumny, gdzie stykają się one z cieczą spływającą w postaci cienkiej warstwy. Dzięki temu zwiększa się powierzchnia zetknięcia pary z cieczą, co wpływa dodatnio na sprawność kolumny. Poza tym, kolumna z wirującą wstęgą wykazuje małą skłonność do zalewania i matę jest orosienie kolumny, co jeszcze bardziej zwiększa jej sprawność. Następna zaleta wirującej wstęgi polega na ułatwionym przelocie par przez kolumnę. Zmniejsza to różnicę ciśnień, która występuje zawsze pomiędzy obszarem szczytowym (ciśnienie mniejsze) i obszarem u dołu kolumny destylacyjnej (ciśnienie większe). Różnica ciśnień zależy od wymiarów kolumny, rodzaju wypełnienia i szybkości destylacji. Duża różnica ciśnień jest niepożądana, gdyż dla przesunięcia par do głowicy konieczny jest wtedy większy dopływ ciepła do kolby destylacyjnej. W przypadku kolumny z wirującą wstęgą różnica ciśnień wynosi tylko 0,3 hPa (0,23 mm Hg). Dzięki temu kolumna ta jest szczególnie dogodna do destylacji frakcyjnej pod zmniejszonym ciśnieniem. Zestaw do destylacji frakcyjnej z wirującą wstęgą jest produkowany w wymiarach odpowiednich do skali mikro i półmikro (np. l—5 ml), do skali laboratoryjnej (np. 250 ml) oraz do skali półtechnicznej (12 l). Sposób wykonania destylacji jest w zasadzie taki sam jak w przypadku zwykłej kolumny destylacyjnej wypełnionej pierścieniami Fenskego
W trosce o komfort korzystania z naszego serwisu chcemy dostarczac Ci coraz lepsze uslugi. By moc to robic prosimy, abys wyrazil zgode na dopasowanie tresci marketingowych do Twoich zachowan w serwisie. Zgoda ta pozwoli nam czesciowo finansowac rozwoj swiadczonych uslug.
Pamietaj, ze dbamy o Twoja prywatnosc. Nie zwiekszamy zakresu naszych uprawnien bez Twojej zgody. Zadbamy rowniez o bezpieczenstwo Twoich danych. Wyrazona zgode mozesz cofnac w kazdej chwili.